Translational Science
Cory
 Theodore Cory, PharmD, PhD, is an Assistant Professor in the Department of Clinical
Pharmacy. He completed his PharmD at Drake University, and a PhD in the Pharmaceutical
Sciences at the University of Kentucky College of Pharmacy. Following the completion
of his PhD, he completed a post-doctoral fellowship in Clinical Pharmacology at the
Antiviral Pharmacology Laboratory at the University of Nebraska Medical Center, under
the direction of Dean Courtney Fletcher.
Theodore Cory, PharmD, PhD, is an Assistant Professor in the Department of Clinical
Pharmacy. He completed his PharmD at Drake University, and a PhD in the Pharmaceutical
Sciences at the University of Kentucky College of Pharmacy. Following the completion
of his PhD, he completed a post-doctoral fellowship in Clinical Pharmacology at the
Antiviral Pharmacology Laboratory at the University of Nebraska Medical Center, under
the direction of Dean Courtney Fletcher.
Contact:
College of Pharmacy Building Rm. 342
Phone: 901.448.7216
Email: tcory1@uthsc.edu
The Cory laboratory is dedicated to improving outcomes in HIV+ individuals by developing strategies to increase the concentrations of drugs used to fight HIV in cells in locations where drug concentrations may be insufficient to prevent replication of the virus. These sites include the brain, lymph nodes, and secondary lymphoid tissues. While CD4+ T cells are the primary target of HIV, macrophages are infected early, and remain an important infected cell population. These two host cells interact in lymph nodes and secondary lymphoid tissue. Macrophages exist in two phenotypically dissimilar polarized subsets, the classically activated (M1) phenotype, which is pro-inflammatory and involved in the destruction of intracellular pathogens, and the alternatively activated (M2) phenotype, which is anti-inflammatory and involved in tissue repair. The role of these two subsets of macrophages in HIV is uncertain, as is the disposition of antiretrovirals in the cells. Our goal is to define the mechanisms by which intracellular antiretroviral concentrations are altered in macrophage subsets, and the effect of this on viral replication and spread by focusing on developing strategies to modulate the expression and function of drug transporters on macrophages.
Additionally, we are investigating strategies to increase drug concentrations in the presence of drugs of abuse, including nicotine and alcohol. These two drugs are commonly used in HIV+ individuals, and are strongly associated with worsened outcomes in these individuals.
Projects in our laboratory include assessing the expression of drug efflux transporters in polarized macrophages, including P-glycoprotein and MRP1, and how these alterations effect the intracellular concentrations of antiretrovirals and viral production from infected cells.
Additionally, we assess how nicotine or ethanol use alters transporter expression in polarized macrophages. Both alcohol and nicotine are commonly abused by individuals with HIV, and are associated with worsened outcomes in these individuals. These is a high importance to determine strategies to improve outcomes in individuals with HIV who either smoke or abuse alcohol.
- Cory, TJ, He H, Winchester LC, Kumar S, Fletcher CV. Alterations in P-Glycoprotein Expression and Function between Macrophage Subsets. Pharmaceutical Research. 2016:33(11):2713-2721.
- Cory TJ, Midde NM, Rao PSS, Kumar S. Investigational Reverse Transcriptase Inhibitors for the Treatment of HIV. Expert Opinion on Investigational Drugs. 2015:24(9):1219-1228.
- Cory TJ, Winchester L, Robbins BL, Fletcher CV. A Rapid Spin Through Oil Results in Higher Cell-Associated Concentrations of Antiretrovirals Compared with Conventional Cell Washing. 2015:7(12):1447-1455.
- Cory TJ, Schacker TW, Stevenson M, Fletcher CV. Overcoming Pharmacologic Sanctuaries. Current Opinion in HIV and AIDS. 2013:8(3):190-195. PMC3677586
Graduate Student: Ying Mu
Collaborator: Santosh Kumar
Fortwendel
Aspergillus fumigatus is among the most common causes of human fungal infection in immunocompromised individuals,
including solid organ transplant recipients, those undergoing hematopoietic stem cell
transplant, and patients receiving highly immunosuppressive chemotherapies. It is
estimated that between 200,000 and 400,000 cases of invasive aspergillosis (IA) occur
annually. If untreated, these infections are almost always fatal, and even with proper
diagnosis and treatment, are associated with an overall 50% mortality rate. Furthermore,
the estimated annual cost of these invasive Aspergillus infections in the U.S. approaches $1 billion.
In the non-immune suppressed patient, Aspergillus species can cause chronic, non-invasive infections that range from asymptomatic colonization
of pre-formed cavitary lesions to inflammatory forms of disease. The inflammatory
disease states, together known as Chronic Pulmonary Aspergillosis (CPA), are recently
recognized by new diagnostic criteria and are actually a collection of syndromes known
as chronic necrotizing, chronic cavitary and chronic fibrotic pulmonary aspergillosis
depending on clinical manifestations. Prior mycobacterial infections, COPD and additional
chronic lung complications are all major predisposing conditions for development of
CPA, conditions that are often further complicated by the presence of the fungus.
CPA is now considered a major under-recognized disease. Therapy options are extremely
limited for the aspergilloses. Resistance to the triazole class of antifungals, the
major class with anti-Aspergillus activity, is on the rise. Although more than a decade of research has focused on characterizing
the emerging threat of triazole resistance in A. fumigatus, strategies for preventing or circumventing this increasingly grave phenomenon remain
elusive.
Our work addresses multiple questions directed at significant knowledge gaps concerning
the elucidation of: 1) host-pathogen interactions during invasive and chronic fungal
diseases; 2) molecular mechanisms of A. fumigatus pathogenic fitness; and 3) and mechanisms of triazole antifungal resistance in Aspergillus species.
Jarrod R. Fortwendel, PhD
Assistant Professor
Department of Clinical Pharmacy and Translational Science
College of Pharmacy
811 Madison Ave,
Memphis, TN 38163
Labs: Room 325/327
Office: Room 343
Phone: 901-448-5140
Email: jfortwen@uthsc.edu
1. Souza ACO, Martin-Vicente A, Nywening AV, Ge W, Lowes DJ, Peters BM, and Fortwendel JR. Loss of Septation Initiation Network (SIN) kinases blocks tissue invasion and unlocks echinocandin cidal activity against Aspergillus fumigatus. PLoS Pathog. 2021; Aug 9;17(8):e1009806. doi: 10.1371/journal.ppat.1009806
2. Pérez-Cantero A, Martin-Vicente A, Guarro J, Fortwendel JR, and Capilla J. Analysis of the cyp51 genes contribution to azole resistance in Aspergillus section Nigri with the CRISPR-Cas9 technique. Antimicrob. Agents Chemother. 2021; Mar 8;65(5):e01996-20. doi: 10.1128/AAC.01996-20.
3. Zhao S, Ge W, Watanabe A, Fortwendel JR and Gibbons JG. Genome-Wide Association for Itraconazole Sensitivity in Non-resistant Clinical Isolates of Aspergillus fumigatus. Front. Fungal Biol. 2021; 1:617338. doi: 10.3389/ffunb.2020.617338.
4. Martin-Vicente A, Souza ACO, Nywening AV, Ge W, and Fortwendel JR. Overexpression of the Aspergillus fumigatus Small GTPase, RsrA, Promotes Polarity Establishment during Germination. J Fungi (Basel). 2020; 6(4):285. doi: 10.3390/jof6040285.
5. Nywening AV, Rybak JM, Rogers PD, and Fortwendel JR. Mechanisms of Triazole Resistance in Aspergillus fumigatus. Environ Microbiol. 2020. doi: 10.1111/1462-2920.15274
6. Esquivel BD, Rybak JM, Barker KS, Fortwendel JR, Rogers PD, and White TC. Characterization of the Efflux Capability and Substrate Specificity of Aspergillus fumigatus PDR5-likeABC Transporters Expressed in Saccharomyces cerevisiae. mBio. 2020. doi: 10.1128/mBio.00338-20.
7. Souza ACO, Al Abdallah Q, DeJarnette K, Martin-Vicente A, Nywening AV, DeJarnette C, Sansevere EA, Ge W, Palmer GE and Fortwendel JR. Differential Requirements of Protein Geranylgeranylation for the Virulence of Human Pathogenic Fungi. Virulence. 2019. 10(1): 511-526. doi: 10.1080/21505594.2019. 1620063.
8. Rybak JM, Ge W, Wiederhold NP, Parker JE, Kelly SL, Rogers PD, and Fortwendel JR. Mutations in hmg1, Challenging the Paradigm of Clinical Triazole Resistance in Aspergillus fumigatus. MBio. 2019. 10(2). pii: e00437-17. doi: 10.1128/mBio.00437-19.
9. Martin-Vicente A, Souza ACO, Al Abdallah Q, Ge W and Fortwendel JR. SH3-class Ras Guanine Nucleotide Exchange Factors are essential for Aspergillus fumigatus invasive growth. Cell. Microbiol. 2019. [Epub Ahead of Print]. Jan 30:e13013. doi: 10.1111/cmi.13013.
10. Rybak JM, Fortwendel JR and Rogers PD. Emerging threat of triazole-resistant Aspergillus fumigatus. J. Antimicrob. Chemother. 2019. 74(4):834-842. doi: 10.1093/jac/dky517.
Adela Martin-Vicente, PhD, Postdoctoral Fellow
Xabier Guruceaga, PhD, Postdoctoral Fellow
Ashley Nywening, Graduate Student (Microbiology, Immunology, & Biochemistry)
Harrison Thorn, Graduate Student (Pharmaceutical Sciences)
Jinhong Xie, Graduate Student (Pharmaceutical Sciences)
Hohmeier
The overarching long-term goal of the Hohmeier lab is to advance the practice of community pharmacy in order to improve patient access to quality, cost-effective health care. My interest in this area is driven by experience as a community pharmacist who developed and delivered niche clinical patient care services based on the unique needs of the community, its patients, and other health care providers. As the most accessible health care professionals, community pharmacists represent a highly trained, clinically-minded workforce capable of scaling-up high quality, evidence-based population health solutions in a cost-effective manner.
Community pharmacy’s impact on public and population health is best illustrated by its contributions to U.S. vaccination rates over the past decade. It was only in 2010 that every state in the U.S. had enacted statutory changes allowing for pharmacists to administer influenza vaccinations. Correspondingly, influenza vaccinations in community pharmacies increased from 3.2 million in 2007 to 20.9 million in 2013. This is of great importance, as rapid implementation of evidence-based interventions across heterogeneous settings and patient populations such as this are uncommon in the health care industry. Therefore, it is our hypothesis that the community pharmacist represents a means to increase the implementation of other evidence-based quality health care interventions in a scalable, widespread, and cost-effective manner.
My research has identified that implementation of novel patient care services in the community pharmacy setting can be best facilitated by both optimizing the role of pharmacist-extenders (such as certified pharmacy technicians) and enhancing electronic communication between pharmacists and other providers. To this end, my community pharmacy research laboratory has undertaken several concurrent projects investigating interventions that aim to: 1) bridge the communication gap between the community pharmacy and other health care facilities, 2) investigate novel roles for pharmacy support staff, and 3) evaluate pharmacy support staff training and credentialing practices and their impact on patient care delivery in the community pharmacy.
Preliminary findings from my ongoing research suggests that it is feasible to integrate a regional health information exchange (HIE) within community pharmacy workflow, and that such HIE integration can facilitate both the development of novel clinical programs, as well as improve the quality and efficiency of the drug utilization review (DUR) process of medication distribution.
Related to pharmacy-extenders, we have uncovered new roles for the pharmacy technician in the delivery of clinical services in the community pharmacy based on work done at high-performing medication therapy management (MTM) centers in Tennessee. Lastly, we are currently investigating technician product verification (TPV) and its impact on the development and sustainability of novel clinical, evidence-based patient care services in the community pharmacy.
Lastly, cost-effective implementation of high quality, evidence-based clinical care is possible in the community pharmacy setting because it has the ability to operate in a consumer-driven manner, similar to convenient care clinics, optometry practices, and dentistry practices. According to my research, their accessibility and extended hours of operation are highly valued traits – especially among younger health care consumers between the ages of 20-35.
Key Collaborators
Justin D. Gatwood, PhD
Nancy B. Hart, PharmD
Chelsea Phillips Renfro, PharmD
James S. Wheeler, PharmD
|
Community Pharmacy Transformation Pilot Project: Optimizing Patient Care by Increasing Community Pharmacist-Provided Care |
National Association of Chain Drug Stores (NACDS)
|
|
Exploring the Effects and Outcomes of Pharmacy Technician Certification Board (PTCB) Certification of Pharmacy Technicians (Phase I and II) |
Pharmacy Technician Certification Board (PTCB) |
|
Consumer Perceptions of Point-of-care Testing |
Community Pharmacy Foundation
|
|
Operation Access: A Four- Part Effort to Expand Access to Community Pharmacy Services Across the State of Tennessee |
NACDS Foundation Pharmacy Partners Grant
|
|
Exploring Current Practices of the Role of the Pharmacy Technician in the Provision of Cognitive Pharmaceutical Services at High Performing Sites within a Chain Community Pharmacy |
NACDS Foundation |
|
Evaluating Hospital Readmissions After Initiating Community Pharmacist- Directed Transitions of Care Program |
APhA Foundation Incentive Grant |
- Hohmeier KC, Boldin S, Spivey CA, Chisholm-Burns MA. Use of Electronic Health Records by Community Pharmacists in Establishing a Transitions of Care Program. J Am Pharm Assoc (2003). 2017;57(5):608-15.
- Hohmeier KC, Loomis B, Gatwood J. Consumer Perceptions of and Willingness-to-Pay for Point-of-Care Testing Services in the Community Pharmacy. Res Social Admin Pharm. 2017;13(4):746-53.
- Hohmeier KC, McDonough SL, Wang J. Co-creation of market expansion in point-of-care testing in the United States: Industry leadership perspectives on the community pharmacy segment. Res Social Admin Pharm. 2017;13(4):746-53
- Justis L, Crain J, Marchetti L, Hohmeier KC. The Effect of Community Pharmacy Technicians on Industry Standard Adherence Performance Measures After Cognitive Pharmaceutical Services Training. J Pharm Technol. 2016;32(6):230-3.
- White CL, Hohmeier KC. Pharmacy Informatics Current and Future Roles for the Pharmacy Technician. J Pharm Technol. 2015;31:247-252.
- Powers MF, Hohmeier KC. Pharmacy Technicians and Immunizations. J Pharm Technol.2011; 27:111-6.
- Hohmeier KC, Baker P, Storey C, Martin N, Gatwood JD. Exploring the Membership Pharmacy Model: Initial impact and feasibility. Journal of the American Pharmacists Association. 2022 Oct 22.
Kumar
CURRENT POSITION AND ADDRESS
Professor, Department of Pharmaceutical Sciences
Assistant Dean for Scholarly Integration and Collaboration
College of Pharmacy
University of Tennessee Health Science Center
881 Madison Ave.
Memphis, TN 38163
Phone: 901-448-7157 (Office)
Phone: 409-370-1664 (Cell)
Email: ksantosh@uthsc.edu
Dr. Santosh Kumar is Assistant Dean for Scholarly Integration and Collaboration and professor in the Department of Pharmaceutical Sciences in the College of Pharmacy at the Universitiy of Tennessee Health Science Center in Memphis. Dr. Kumar graduated from the Indian Institute of Technology (IIT)-Bombay, India. He did his post-doctorate fellowship from the University of Missouri-Kansas City (UMKC) followed by worked as a junior faculty at the University of Texas Medical Branch. Dr. Kumar then went back to UMKC where he worked as an Assistant Professor before coming to UTHSC in 2014. Dr. Kumar is a trained biochemist and enzymologist with expertise in drug metabolism, HIV, substance abuse, and exosomes. Dr. Kumar’s group has published substantially (130 papers). His research has been funded by various NIH grants including R01 and R21. Dr. Kumar has mentored six graduate students and three post-doctorate fellows along with faculty members and numerous other trainees. Currently, he is mentoring three graduate students, two PDFs, and two faculties. In addition to research, Dr. Kumar participate significantly in classroom teaching to both professional pharmacy students and graduate students. Dr. Kumar has been actively engaged in serving the Society on Neuroimmune Pharmacology at various executive levels including President-elect. As a result of his distinguished contributions to research, teaching, mentoring, and service, Dr. Kumar has received numerous awards and honors.
Lab/Research Description
Our research program is at the intersection of HIV-1, drugs of abuse, cytochrome P450, and exosomes. Drugs of abuse, especially alcohol drinking and tobacco smoking are highly prevalent in HIV-infected individuals. Alcohol and tobacco are known to augment HIV replication and reduce the response to antiretroviral therapy leading to exacerbated HIV pathogenesis and AIDS/NeuroAIDS. However, the mechanisms by which these phenomena occur are largely unknown. We propose that alcohol- and tobacco-mediated increases in HIV replication and decreases in the response to antiretroviral therapy occur through the cytochrome P450 (CYP) pathway. Our ongoing projects are to study the “role of CYP in alcohol- and tobacco-mediated HIV-1 pathogenesis and antiretroviral therapy”. The alcohol project is funded by NIH/NIAAA. Our group is the first one to show a potential role of CYP pathways within the context of drugs of abuse-mediated HIV pathogenesis. This provides a novel target to treat HIV-infected drug abusers effectively.
- Alcohol, HIV, antiretroviral therapy (ART), and CYP: Since alcohol and many ARTs such as non-nucleoside reverse transcriptase inhibitors (NNRTIs) and protease inhibitors (PIs) are metabolized by the CYP family of enzymes, causing oxidative stress and drug-drug interactions, our group developed a research niche in alcohol and HIV. Alcohol use is highly prevalent in HIV populations, and it is known to increase HIV replication and decrease response to ART drugs. In this project, our group has shown the1) potential involvement of CYP pathways in alcohol-PI interactions, which may alter efficacy of ART and ART-mediated toxicity in HIV-infected individuals, 2) mechanism by which CYP2E1 (an alcohol-metabolizing enzyme) is regulated by ethanol in monocytic and astrocytic cell lines, 3) potential role of CYP and oxidative stress pathways in alcohol-mediated HIV pathogenesis using in vitro models and ex-vivo human subjects, and 4) role of CYP enzymes in alcohol-mediated effects on bioavailability of protease and integrase inhibitors.
- Tobacco/nicotine, HIV, and CYP: Since smoking constituents such as nicotine and polyaromatic hydrocarbons (PAHs) are metabolized by the CYP family of enzymes causing oxidative stress, we developed a research niche in tobacco/nicotine and HIV. Smoking is highly prevalent in HIV populations, and it is known to increase HIV replication. In this project, our laboratory has shown the: 1) potential involvement of CYP pathways in nicotine-mediated oxidative stress in HIV systems, monocytic and astrocytic cell lines, 2) role of CYP-mediated nicotine metabolism and oxidative stress pathways in HIV replication using an ex-vivo study that utilized HIV-infected smokers, 3) potential involvement of cigarette smoke condensate (CSC) and PAHs, especially benzo(a)pyrene, in smoking mediated oxidative stress and cellular toxicity, perhaps through CYP pathways that may lead to HIV pathogenesis.
- Exosomes, HIV, drugs of abuse, and exosomes. Exosomes are clinically relevant in developing biological markers and novel therapeutics for many diseases. The known role of exosomes in the context of drugs of abuse and HIV is poorly understood. We propose that exosomes derived from macrophages and microglia play an important role in drugs of abuse, especially smoking- and alcohol- mediated effects on HIV pathogenesis and neuronal damage. We also propose that exosomal CYPs, antioxidant and pro-oxidant enzymes, and cytokines, as well as miRNAs that regulate these enzymes, play important role in such effects. This project is funded by NIH/NIDA R21. In this project, we are investigating the potential role of exosomal CYP, AOE, and cytokines in smoking-mediated cytotoxicity and HIV-1 pathogenesis. Further, we plan to identify and characterize the specific contents (e.g. protein, cytokine, miRNA, metabolic contents) in exosomes which might be responsible for tobacco-exacerbated HIV replication and neuronal damage that have clinical relevance to neuro-AIDS. In this project, we also plan to examine the potential role of exosomal enzymes (cathepsins B and D) in mediating alcohol-induced neurotoxicity.
- Exosomes, involvement of CYP2E1 in alcohol induced neurotoxicity: This Project was developed based on our pilot study findings where plasma exosomes of a healthy population contained a high level of metabolically active CYP2E1 compared to the other CYPs. Moreover, hepatic cells secrete exosomes at a higher rate under stress conditions such as exposure to ethanol (ETH). Therefore, we propose that translocation of circulating plasma exosomes and their CYP2E1 cargo can potentially exacerbate the alcohol-induced cytotoxicity in extra-hepatic tissues such as neuronal cells.
- CYP2E1 inhibition and drug-induced toxicity: We also work in the area of drug discovery where we aim to find better analogs of diallyl sulfide (DAS) which will retain the CYP2E1 inhibition property without causing significant drug induced cellular toxicity. In this project, we have shown that: 1) efficacies of DAS analogs are equivalent to that of DAS in inhibiting CYP2E1, 2) DAS analogs have better acute and chronic safety profiles than DAS and 3) DAS analogs can rescue cells from ethanol- and acetaminophen-mediated toxicity. We proposed to validate the PK of these analogs in animal model.
- Antiretroviral therapy (ART) and nanoformulations: Monocytes serve as sanctuary sites for HIV from which the virus is difficult to be eliminated. Therefore, an effective ART strategy is critical for effective viral suppression in monocytes. This study focuses on a new strategy using nanoformulation to optimize the efficacy of ART drugs in HIV-infected monocytes, with the ability to cross the blood-brain barrier to improve HIV treatment outcomes. In this project: 1) we developed a PLGA-EVG nanoparticle formulation which demonstrated time- and concentration-dependent uptakes in monocytes and superior viral suppression for a prolonged period of time in HIV-infected macrophages. We are now in the process of using these nanoparticles to specifically target monocytes and to examine their role in infiltrating the BBB and suppressing HIV in CNS cells.
Members
- Dr. Santosh Kumar (Principal Investigator)
- Mrs. Namita Sinha (Laboratory Supervisor)
- Sunitha Kodidela (Post-doctoral fellow)
- Sabina Ranjit (Graduate research assistant)
- Mohammad Arifur Rahman, (Graduate research assistant)
- Benjamin Patters (Graduate research assistant)
- Yuqing Gong (Graduate research assistant)
- Sanjana Haque (Graduate research assistant)
- Kelli Gerth (Pharm D/PhD student)
Past Members
- Paluri Sai Shantanu Rao (Research Associate): Assistant Professor, College of Pharmacy, The University of Findley, Findley, OH
- Narasimha Midde (Research Associate): a=Associate Director, KinderPharm LLC, Exton, PA
Collaborators
- Theodore Cory, Assistant Professor, Department of Clinical Pharmacy, College of Pharmacy, University of Tennessee Health Science Center
Title: HIV, smoking, alcohol, and antiretroviral
- Wei Li, Professor, Department of Pharmaceutical Sciences, College of Pharmacy, University of Tennessee Health Science Center
Title: Synthesis and characterizations of novel analoges of diallyl sulfide
- Fatah Kashanchi, Professor, Laboratory of Molecular Virology, George Mason University
Title: HIV and substance abuse, exosomes, and oxidative stress
- Hao Chen, Associate Professor, Department of Pharmacology, Addiction Science, and Toxicology, University of Tennessee Health Science Center
Title: Nicotine-HIV interactions and neuroinflammation
|
1. Kumar S. (2021). Finding an effective way to create learning environments for didactic courses
in a virtual classroom setting. Pharmacy Education, 21, 621–625. https://doi.org/https://doi.org/10.46542/pe.2021.211.621625 |
Laizure-Parker
 The Laizure-Parker lab focuses on understanding mechanisms affecting the variability
in metabolism and disposition of drugs metabolized by the human carboxylesterase (CES)
enzymes. In humans, two carboxylesterases, carboxylesterase-1 (CES1) and carboxylesterase-2
(CES2), found primarily in the liver and intestine respectively, play an important
role in the biotransformation of a growing number of widely prescribed medications
used to treat many common disorders (Table).
The Laizure-Parker lab focuses on understanding mechanisms affecting the variability
in metabolism and disposition of drugs metabolized by the human carboxylesterase (CES)
enzymes. In humans, two carboxylesterases, carboxylesterase-1 (CES1) and carboxylesterase-2
(CES2), found primarily in the liver and intestine respectively, play an important
role in the biotransformation of a growing number of widely prescribed medications
used to treat many common disorders (Table).
Factors affecting the CES activity would be expected to markedly alter the pharmacokinetics and clinical effects of substrate drugs. One key factor that could affect catalytic activity is drug interactions that inhibit CES function. The importance of inhibition of drug metabolism in medication safety and efficacy is well established for drugs that undergo metabolism by cytochrome P450 enzymes. In distinct contrast, little is known about the potential for CES to serve as a target for metabolic inhibition mediated by drug interactions.
 How drug-induced metabolic inhibition affects the safety and effectiveness of CES
substrate drugs is a major gap in our understanding of adverse drug events. During
the last 20 years there has been an increase of prescribed CES substrate drugs; however
our knowledge of the mechanisms, clinical importance and toxicity of drug interactions
with these enzymes has yet to advance beyond in vitro metabolic studies. We are interested
in exploring how inhibition of CES by drug interactions affects the metabolism, disposition,
and response to medications hydrolyzed by these enzymes.
How drug-induced metabolic inhibition affects the safety and effectiveness of CES
substrate drugs is a major gap in our understanding of adverse drug events. During
the last 20 years there has been an increase of prescribed CES substrate drugs; however
our knowledge of the mechanisms, clinical importance and toxicity of drug interactions
with these enzymes has yet to advance beyond in vitro metabolic studies. We are interested
in exploring how inhibition of CES by drug interactions affects the metabolism, disposition,
and response to medications hydrolyzed by these enzymes.
 To address these questions, our laboratory uses a combination of in vitro metabolic
studies, in vivo models, human studies, and modeling. We have identified a number
of potent CES inhibitors including alcohol (ethanol), diltiazem, verapamil, aripiprazole,
loperamide, and rivastigmine.
To address these questions, our laboratory uses a combination of in vitro metabolic
studies, in vivo models, human studies, and modeling. We have identified a number
of potent CES inhibitors including alcohol (ethanol), diltiazem, verapamil, aripiprazole,
loperamide, and rivastigmine.
Contact Info:
University of Tennessee Health Science Center
Department of Clinical Pharmacy
College of Pharmacy
881 Madison Ave.,
Memphis, TN 38163
Lab: Room 311; Office: Room 346 (Parker) and Laizure (358)
Phone: 901-448-7143 (Parker) and 901-448-6310 (Laizure)
Email: rparker@uthsc.edu, claizure@uthsc.edu
Center for Pediatric Pharmacokinetics and Therapeutics
Dr. Feng Chen (Postdoctoral fellow)
Palmer
 An estimated 1.5 million people die each year from invasive fungal infections, and
many millions more are afflicted by debilitating mucosal and subcutaneous mycoses.
Current antifungal therapies have serious deficiencies including poor efficacy, limited
spectrum of activity, patient toxicity and the emergence of resistant fungi. Consequently,
mortality rates are disturbingly high. A major obstacle to developing effective new
antifungal drugs is the fundamental similarity between the cells of these eukaryotic
pathogens and their mammalian host. This presents a challenge in devising therapeutic
agents with pathogen selective toxicity. A major long-term goal of my research program
is to identify and validate new target proteins that can provide a basis to develop
efficacious new antifungal therapies. Current investigations within my lab include
the discovery and development of new classes of antifungal agents that target either:
1). The integrity of a sub-cellular organelle called the fungal vacuole; 2). Fungal
fatty acid biosynthesis; and 3) aromatic amino acid biosynthesis. As part of these
studies we have devised several high-throughput (HTP) chemical screening assays to
identify compounds that target these cellular functions. This includes a new and broadly
applicable type of target based whole-cell screen (TB-WCS) that combines the benefits
of both traditional target-based and cell-based chemical screens into a single HTP
assay. We anticipate our TB-WCS approach to chemical screening will greatly enhance
the speed and efficiency with which new pre-therapeutic leads, with a defined mechanism
of action can be identified. Through these efforts, I have become increasingly excited
about the enormous potential of applying yeast based systems (which are highly amenable
to HTP approaches) to the discovery of new pharmacotherapies that target human disease
related proteins.
An estimated 1.5 million people die each year from invasive fungal infections, and
many millions more are afflicted by debilitating mucosal and subcutaneous mycoses.
Current antifungal therapies have serious deficiencies including poor efficacy, limited
spectrum of activity, patient toxicity and the emergence of resistant fungi. Consequently,
mortality rates are disturbingly high. A major obstacle to developing effective new
antifungal drugs is the fundamental similarity between the cells of these eukaryotic
pathogens and their mammalian host. This presents a challenge in devising therapeutic
agents with pathogen selective toxicity. A major long-term goal of my research program
is to identify and validate new target proteins that can provide a basis to develop
efficacious new antifungal therapies. Current investigations within my lab include
the discovery and development of new classes of antifungal agents that target either:
1). The integrity of a sub-cellular organelle called the fungal vacuole; 2). Fungal
fatty acid biosynthesis; and 3) aromatic amino acid biosynthesis. As part of these
studies we have devised several high-throughput (HTP) chemical screening assays to
identify compounds that target these cellular functions. This includes a new and broadly
applicable type of target based whole-cell screen (TB-WCS) that combines the benefits
of both traditional target-based and cell-based chemical screens into a single HTP
assay. We anticipate our TB-WCS approach to chemical screening will greatly enhance
the speed and efficiency with which new pre-therapeutic leads, with a defined mechanism
of action can be identified. Through these efforts, I have become increasingly excited
about the enormous potential of applying yeast based systems (which are highly amenable
to HTP approaches) to the discovery of new pharmacotherapies that target human disease
related proteins.
Glen E. Palmer
Associate Professor, Clinical Pharmacy
PhD Genetics, University of Leicester, United Kingdom
BSc. Genetics, University of Sheffield, United Kingdom
Email: gpalmer5@uthsc.edu
Office: 901.448.3744
Lab manager - Tracy Peters M.S.
Postdoctoral researchers – Helene Tournu Ph.D; Arielle Butts Ph.D; Arturo Luna-Tapia
Ph.D
Research Associate – Kathy Barker Ph.D
Graduate Students – Christian DeJarnette B.S.; Parker Reitler B.S.
- Tournu, H., Carroll, J., Latimer, B., Dragoi, A., Dykes, S., Cardelli, J., Peters, T.L., Eberle, K.E., and Palmer, G.E. (2017). Identification of small molecules that disrupt vacuolar function in the pathogen Candida albicans. Accepted for publication in PLoS One.
- Luna-Tapia, A., Peters, B.M., Tournu, H., and Palmer, G.E. (2017). An azole tolerant endosomal trafficking mutant of Candida albicans is susceptible to azole treatment in a mouse model of vaginal candidiasis. Accepted for publication in Antimicrobial Agents and Chemotherapy.
- Luna-Tapia, A., Kerns, M., Eberle, K.E., Jursic, B.S., and Palmer, G.E. (2015). Trafficking through the late endosome significantly impacts Candida albicans tolerance of the azole antifungals. Antimicrobial Agents and Chemotherapy, 59: 2410-20. PMCID:
- Luna-Tapia, A., Tournu, H., Peters, T., and Palmer, G.E. (2016). Endosomal trafficking defects can induce calcium dependent azole tolerance in Candida albicans. Antimicrobial Agents and Chemotherapy, Sep 19: AAC.01034-16. [Epub ahead of print].
- Luna-Tapia, A., Peters, B.M., Eberle, K.E., Kerns, M.E., Foster, T.P., Marrero, L., Noverr, M.C., Fidel, P.L. Jr, Palmer, G.E. (2015). ERG2 and ERG24 are required for normal vacuolar physiology as well as Candida albicans pathogenicity in a murine model of disseminated but not vaginal candidiasis. Eukaryotic Cell, 14: 1006-1016. PMCID:
Peters
The Peters lab has two main foci of research: 1) the host and fungal molecular mechanisms responsible for the immunopathogenesis of
vulvovaginal candidiasis and 2) quorum sensing and toxin regulation during fungal-bacterial intra-abdominal infection.
For more information visit: https://www.thepeterslab.com
Current Members:
Jennifer Carnahan, BS (Lab Manager / Researcher I)
Gustavo Alvira-Arill, PharmD (Pediatric Infectious Diseases Pharmacy Fellow)
Saikat Paul, PhD (Postdoctoral Fellow)
Jian Miao, MS (PhD Student, Pharmaceutical Sciences)
Amirhossein Davari, MS (PhD Student, Pharmaceutical Sciences)
Previous Members:
Olivia Todd, PhD (currently postdoctoral fellow at Univ. of Chicago)
Marjoleine Willems, PhD (currently Staff Scientist at Univ. of Georgia)
Emily Sansevere, PhD (currently Translational Medicine Lead at Finch Therapeutics)
Junyan Liu, PhD (currently Assistant Professor, SCUT and Ministry of Agriculture key
laboratory)
Zhenbo Xu, PhD (currently Associate Professor at SCUT)
Kathy Barker, PhD (currently Senior Scientist at St. Jude)
David Lowes, PhD (currently in industry)
Molly Coltrane, PharmD (pharmacy resident at NYU Winthrop)
Amanda Vogel, MS (recent MS graduate)
Winter Bruner, BS (currently PhD candidate at UTHSC)
Jabez Fortwendel (currently completing BS at UTK)
Key Collaborators:
Jeremy Stultz, PharmD (UTHSC)
Mairi Noverr, PhD (LSUHSC)
Paul Fidel, PhD (LSUHSC)
Glen Palmer, PhD (UTHSC)
Jarrod Fortwendel, PhD (UTHSC)
Sarah Gaffen, PhD (Univ. Pittsburgh)
David Williams, PhD (ETSU)
Mike Kruppa, PhD (ETSU)
Vinnie Bruno, PhD (UMB)
Fran Barrera, PhD (UTK)
Joe Pierre, PhD (Univ. Wisconsin)
Jim Cassat, MD, PhD (Vanderbilt)
Candida albicans, an opportunistic human fungal pathogen, is the leading causative agent of vulvovaginal candidiasis (VVC) and presents major quality of life issues for women worldwide. It is estimated that nearly every woman of childbearing age will be afflicted by VVC at least once in her lifetime. Although these treatments are typically effective at reducing organism burden, static function of azole activity, fungal recalcitrance to clearance, and lack of comprehensive understanding of disease pathology necessitates further insight into the host and fungal factors that contribute to vaginitis immunopathology.
[1] We are interested in exploring virulence mechanisms utilized by C. albicans, including the fungal toxin Candidalysin, to activate inflammasome signaling at the vaginal mucosa. We are also focused on determining the downstream signaling events relevant to disease pathogenesis, including the protective role of innate IL-17 signaling at the vaginal mucosa. [2] We are also currently testing sulfonylurea drugs as repurposed adjunctive therapeutic agents to more quickly arrest symptomatic disease.
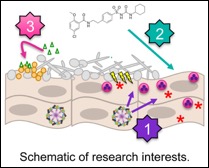 Polymicrobial intra-abdominal infection:
Polymicrobial intra-abdominal infection:
[3] Microorganisms rarely exist as single species communities but instead exist within multi-species consortia where mutually beneficial, parasitic, and antagonistic interactions may develop. However, relatively little is known about the functional consequences of these interactions as they relate to health and disease.
We aim to determine the complex inter-microbial signaling events that mediate infectious synergism observed during intra-abdominal infection with the ubiquitous bacterial pathogen Staphylococcus aureus and the fungus C. albicans. Current studies are focused on identifying activation of the S. aureus agr-quorum sensing system and downstream toxins as key pathways contributing to lethal infection. This polymicrobial intra-abdominal infection serves as an excellent model system for determining microbe-microbe induced virulence gene regulation in vivo. Identification of virulence determinants may serve as rationale for selection of vaccine candidates to reduce lethality clinically associated with fungal-bacterial intra-abdominal infection.
Current funding:
First Tennessee Endowed Chair of Excellence in Clinical Pharmacy (role: chair)
R01AI134796 (role: PI)
R21AI153768 (role: MPI)
R21DK129890 (role: Co-I)
R21AI163503 (role: consultant)
Selected completed funding:
R21AI141829 (role: PI)
R21AI127942 (role: PI)
R01AI116025 (role: Co-I)
K22AI110541 (role: PI)
Institutional support from the COP, CPET, and UTHSC
Selected publications over five years (2017-present)
For a full publication list see here: http://www.ncbi.nlm.nih.gov/myncbi/1FwrSoadqx1kn/bibliography/48206767/public/?sort=date&direction=ascending
1. Liu J, Vogel AK, Miao J, Carnahan JA, Lowes DJ, Rybak JM, Peters BM. Rapid hypothesis testing in Candida albicans clinical isolates using a cloning-free, modular, and recyclable system for CRISPR-Cas9 mediated mutant and revertant construction. Microbiol Spectr. 2022 May 25:e0263021.
2. Liu J, Willems HME, Sansevere EA, Allert S, Barker KS, Lowes DJ, Dixson AC, Xu Z, Miao J, DeJarnette C, Tournu H, Palmer GE, Richardson JP, Barrera FN, Hube B, Naglik JR, Peters BM. A variant ECE1 allele contributes to reduced pathogenicity of Candida albicans during vulvovaginal candidiasis. PLoS Pathog. 2021 Sep 10;17(9):e1009884.
3. Souza ACO, Martin-Vincente A, Nywenning AV, Ge W, Lowes DJ, Peters BM, Fortwendel JR. Loss of Septation Initiation Network (SIN) kinases blocks tissue invasion and unlocks echinocandin cidal activity against Aspergillus fumigatus. PLoS Pathog. 2021 Aug 9;17(8):e1009806.
4. Lowes DJ, Miao J, Al-waqfi RA, Avad KA, Hevener KE, Peters BM. Identification of dual-target compounds with antifungal and anti-NLRP3 inflammasome activity. ACS Infect Dis. 2021 Aug 13;7(8):2522-2535.
5. Miao J, Willems HME, Peters BM. Exogenous reproductive hormones nor Candida albicans colonization alter the near neutral mouse vaginal pH. Infect Immun. 2021. Jan 19;89(2):e00550-20. *selected as Spotlight article.
6. Willems HME, Ahmed SS, Liu J, Xu Z, .Peters BM. Vulvovaginal Candidiasis: A current understanding and burning questions. J Fungi. 2020 Feb 25;6(1):27.
7. Peters BM, Coleman BM, Willems HME, Barker KS, Aggor FEY, Cipolla E, Verma AH, Bishu S, Huppler AH, Bruno VM, Gaffen SL. The IL-17R/IL-22R signaling axis is dispensable for vulvovaginal candidiasis regardless of estrogen status. J Infect Dis. 2020 Apr 7;221(9):1554-1563.
8. Lowes DJ, Hevener KE, Peters BM. The second generation anti-diabetic sulfonylureas inhibit Candida albicans and Candidalysin mediated activation of the NLRP3 inflammasome. Antimicrob Agents Chemother. 2020 Jan 27;64(2):e01777-19.
9. Willis KA, Purvis JH, Myers ED, Aziz MM, Karabayir I, Gomes CK, Peters BM, Akbilgic O, Talati AJ, Pierre JF. Fungi form interkingdom communities in the primordial human gut that develop with gestational age. FASEB J. 2019 Nov;33(11):12825-12837.
10. Willems HME, Stultz JS, Coltrane ME, Fortwendel JP, Peters BM. Disparate Candida albicans biofilm formation in clinical lipid emulsions due to capric acid mediated inhibition. Antimicrob Agents Chemother. 2019. Aug 12. Epub ahead of print.
11. Todd OA, Fidel Jr. PL, Harro JM, Hilliar JJ, Tkaczyk C, Sellman BR, Noverr MC, Peters BM. Candida albicans augments Staphylococcus aureus virulence by engaging the staphylococcal agr quorum sensing system. mBio. 2019 Jun 4;10(3). pii: e00910-19.
12. Luna-Tapia A*, Willems HME,*, Parker JE, Tournu H, Barker KS, Nishimoto AT, Rogers PD, Kelly SL, Peters BM, Palmer GE. Loss of Upc2p inducible ERG3 transcription is sufficient to confer niche-specific azole resistance without compromising Candida albicans pathogenicity. *denotes authors of equal contribution. mBio. 2018 May 22;9(3).
13. Richardson JP†, Willems HME†, Moyes DL, Shoaie S, Tan SL, Barker KS, Palmer GE, Hube B, Naglik JR*, Peters BM*. Candidalysin drives epithelial signaling, neutrophil recruitment, and immunopathology at the vaginal mucosa. Infect Immun. *, † denotes authors of equal contribution. Nov 2017. ** selected as Spotlight article.